I
Ice-lined refrigerator: A compression cycle refrigerator with an internal lining surrounding the storage that is filled with ice, cold water, or other coolant. When the electricity supply fails, the ice, cold water or coolant keeps the refrigerator cool for a minimum of 20 hours without power. (WHO)
Ice-pack: A water-pack that has been frozen to a temperature between −5.0°C and −25.0°C before use.
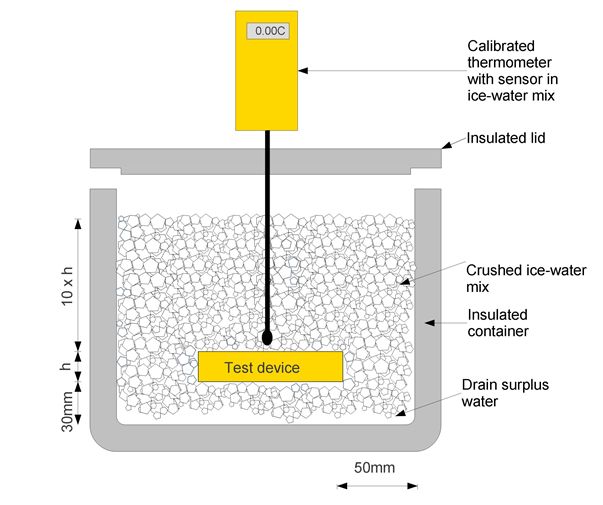
Ice-water bath: A bath of ice and water to maintain a temperature of 0.0°C. The ice-water bath provides an accurate reference temperature at 0.0°C if the melting ice-water mixture is properly set up, handled and maintained. An accurate temperature is achieved by this method because an ice-water mixture in a container which is open to the atmosphere will stabilize at its own “triple point”. At this point all three aggregate states of water coexist: liquid, solid and gaseous. For more physical details refer to ASTM E563-11.
Ice-water bath is a relatively simple method for checking the accuracy of temperature control and monitoring devices. The accuracy of the results is dependent upon the use of a high-quality reference thermometer with a valid calibration certificate. (WHO)
Ice-water bath arrangement for checking the accuracy of a temperature control/monitoring device (WHO)
ICH: International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use - ICH’s mission is to make recommendations towards achieving greater harmonisation in the interpretation and application of technical guidelines and requirements for pharmaceutical product registration, thereby reducing or obviating duplication of testing carried out during the research and development of new human medicines.
Launched in 1990, ICH is a unique undertaking that brings together the drug regulatory authorities and the pharmaceutical industry of Europe, Japan and the United States.
Regulatory harmonisation offers many direct benefits to both regulatory authorities and the pharmaceutical industry with beneficial impact for the protection of public health. Key benefits include: preventing duplication of clinical trials in humans and minimising the use of animal testing without compromising safety and effectiveness; streamlining the regulatory assessment process for new drug applications; and reducing the development times and resources for drug development.
Harmonisation is achieved through the development of ICH Tripartite Guidelines. The Guidelines are developed through a process of scientific consensus with regulatory and industry experts working side-by-side. Key to the success of this process is the commitment of the ICH regulators to implement the final Guidelines.
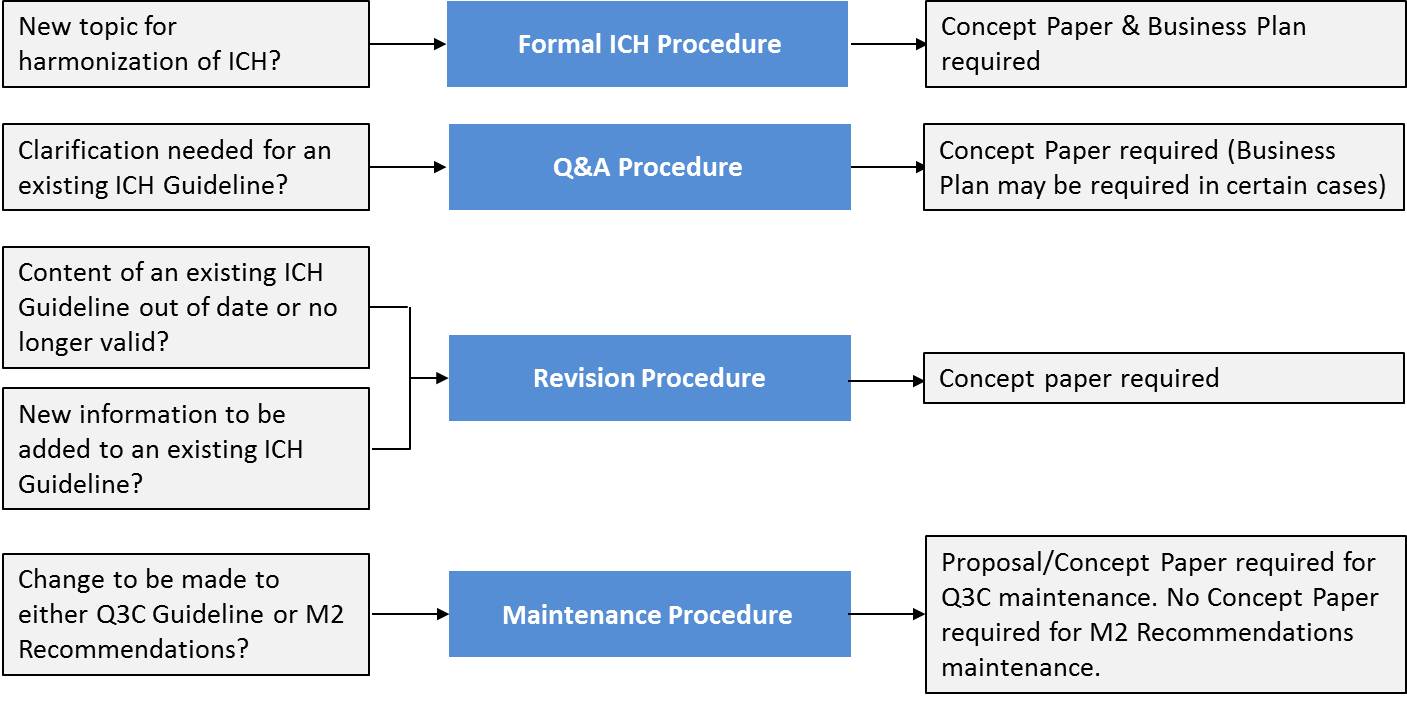
ICH harmonisation activities fall into four categories: Formal ICH Procedure, Q&A Procedure, Revision Procedure and Maintenance Procedure, depending on the activity to be undertaken.
Process of harmonization (ICH)
Immune: Protected from or resistant to a disease or infection by a pathogenic organism as a result of the development of antibodies or cell mediated immunity.
Immunity: The state of being immune to or protected from a disease. That resistance usually associated with the presence of antibodies or cells having a specific action on the microorganism concerned with a particular infectious disease or on its toxin. Effective immunity includes both cellular immunity, which is conferred by T-lymphocyte sensitization, and/or humoral immunity, which is based on B-lymphocyte response. Passive immunity is attained either naturally by transplacental transfer from the mother, or artificially by inoculation of specific protective antibodies (from immunized animals, or convalescent hyperimmune serum or immune serum globulin [human]); it is of short duration (days to months). Active humoral Immunity, which usually lasts for years, is attained either naturally by infection with or without clinical manifestations, or artificially by inoculation of the agent itself in killed, modified or variant form, or of fractions or products of the agent.
Immunity (acquired): Immunity resulting from the development of active or passive immunity, as opposed to natural or innate immunity.
Immunity (herd): Immune protection through vaccination of a portion of the population, which may reduce the spread of a disease by limiting the number of potential hosts for the pathogen. (WHO)
Immunization anxiety-related reaction: An AEFI arising from anxiety about the immunization. (WHO)
Immunization card: A record that contains immunization history and status of a person. The card may be the only record of immunization history and status available for health workers if immunization registers are not well maintained or if clients move from one health facility to another.
Sample immunization card for infants (WHO)

Immunization error-related reaction: (formerly programmatic error) An AEFI that is caused by inappropriate vaccine handling, prescribing or administration and thus, by its nature, is preventable. (WHO)
Immunization register: An official list or record of immunization services they offer to each infant and to pregnant women.
Sample immunization register for infants (WHO)

Immunization safety: The public health practices and policies dealing with the various aspects of the correct administration of vaccines, focusing on minimizing the risk of transmission of disease with the injection and maximizing the effectiveness of the vaccine. The term encompasses the spectrum of events from proper manufacture to correct administration. (WHO)
Immunogenicity: The capacity of a vaccine to induce antibody-mediated and/or cell-mediated immunity and/or immunological memory. (WHO)
Impact indicator: See damage indicator.
Impartial witness: A person, who is independent of the trial, who cannot be unfairly influenced by people involved with the trial, who attends the informed consent process if the subject or the subject’s legally acceptable representative cannot read, and who reads the informed consent form and any other written information supplied to the subject. (ICH E6/R1)
Impermeable containers: Containers that provide a permanent barrier to the passage of gases or solvents, e.g., sealed aluminium tubes for semisolids, sealed glass ampoules for solutions and aluminium/aluminium blisters for solid dosage forms.
An example of impermeable primary container, BCG vaccine from Japan BCG (Kartoglu)

Impurity: Any component of the new drug substance that is not the chemical entity defined as the new drug substance. (ICH Q3A/R2)
Impurity profile: A description of the identified and unidentified impurities present in a new drug substance. (ICH Q3A/R2)
In use: See utilization period.
Incidence: The number of persons who fall ill with a certain disease during a defined time period. (WHO)
Incident: An event or circumstance, which could have or did lead to unintended and/or unnecessary harm to a person, and/or a complaint, loss or damage. (Patient Safety International)
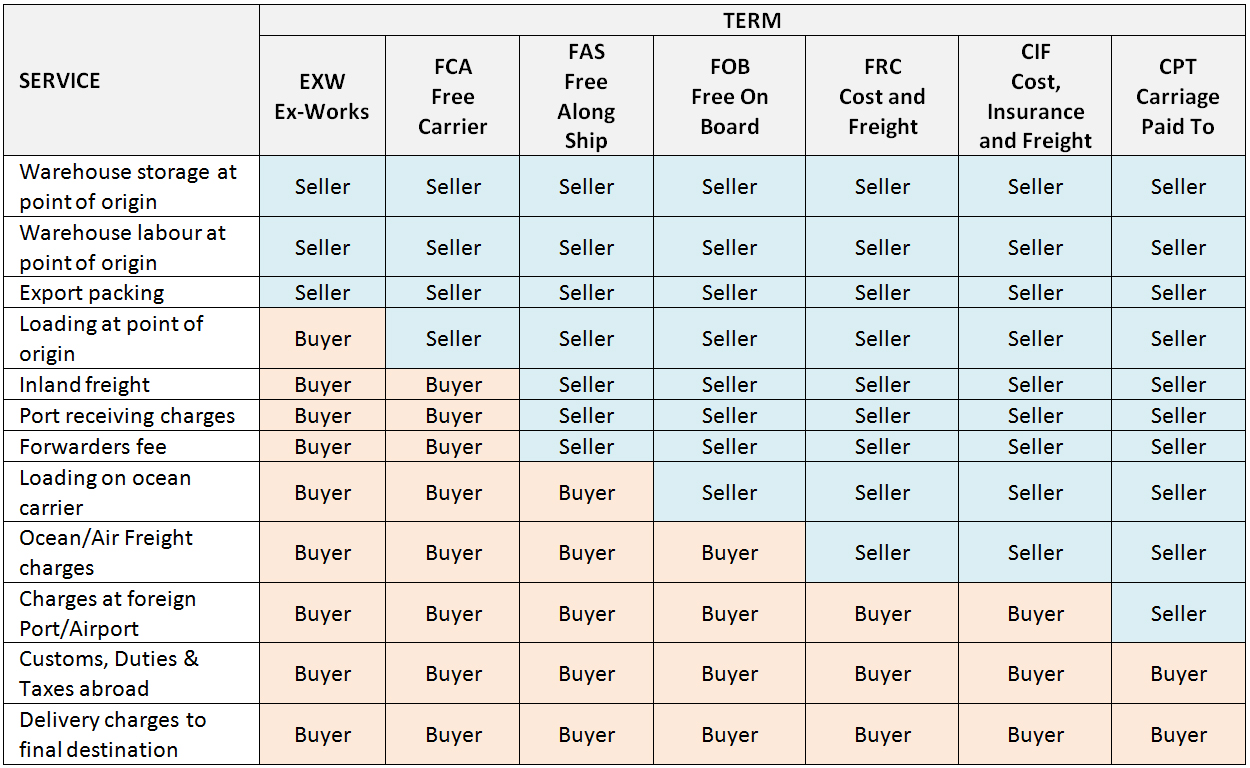
Incoterms: Manufacturers and suppliers offer delivery arrangements based on international standard definitions known as Incoterms. A new edition of Incoterms was published in 2010 and is being phased in. Those below refer to the long-established 2000 edition. It is essential to check which version is being used by the supplier.
Incoterms definitions (WHO)
Independent Data-Monitoring Committee (IDMC): (Data and Safety Monitoring Board, Monitoring Committee, Data Monitoring Committee) An independent data-monitoring committee that may be established by the sponsor to assess at intervals the progress of a clinical trial, the safety data, and the critical efficacy endpoints, and to recommend to the sponsor whether to continue, modify, or stop a trial. (ICH E6/R1)
Independent ethics committee (IEC): An independent body (a review board or a committee, institutional, regional or national), constituted of medical, scientific and non-scientific members, whose responsibility is to ensure the protection of the rights, safety and well-being of human subjects involved in a trial by, among other things, reviewing, approving, and providing continuing review of trial protocol and amendments and of the methods and material to be used in obtaining and documenting informed consent of the trial subjects. Ethics committees should be constituted and operated so that their tasks can be executed free from bias and from any influence of those who are conducting the trial. Such committees are also known variously as Ethics Committee (EC), Institutional Review Board (IRB), Research Ethics Board (REB), Research Ethics Committee, and other designations. ICH E6/R1 Guideline for Good Clinical Practice (current step 4 version, dated 10 June 1996) also defines the roles and responsibilities of IEC. Responsibilities can be summarized as follows:
- Safeguarding the rights, safety, and well-being of all trial subjects (paying special attention to trials that may include vulnerable subjects).
- Reviewing the proposed clinical trial within a reasonable time and document its views in writing, clearly identifying the trial, the documents reviewed and the dates for either approval/favorable opinion; modifications required for its approval; disapproval/negative opinion; and termination/suspension of any prior approval/favorable opinion.
- Considering the qualifications of the investigator for the proposed trial.
- Conducting continuing review of each ongoing trial at intervals appropriate to the degree of risk to human subjects, but at least once per year.
- Reviewing both the amount and method of payment to subjects to assure that neither presents problems of coercion or undue influence on the trial subjects.
- Ensuring that information regarding payment to subjects, including the methods, amounts, and schedule of payment to trial subjects, is set forth in the written informed consent form and any other written information to be provided to subjects.
When a non-therapeutic trial is to be carried out with the consent of the subject’s legally acceptable representative, the IEC should determine that the proposed protocol and/or other document(s) adequately addresses relevant ethical concerns and meets applicable regulatory requirements for such trials. Where the protocol indicates that prior consent of the trial subject or the subject’s legally acceptable representative is not possible, the IEC should determine that the proposed protocol and/or other document(s) adequately addresses relevant ethical concerns and meets applicable regulatory requirements for such trials (i.e., in emergency situations). The IEC may also request more information be given to subjects when, in the judgement of the IEC, the additional information would add meaningfully to the protection of the rights, safety and/or well-being of the subjects.
ICH suggests that the composition of the IEC should include at least five members, with at least one member whose primary area of interest is in a non-scientific area, and at least one member who is independent of the institution/trial site.
Independent variable: Inputs or causes used in an experiment or modelling, or are tested to see if they are the cause. Independent variable is the one that is changed by the experimenter. Independent variable is also known as a “predictor variable”, “regressor”, “controlled variable”, “manipulated variable”, and “explanatory variable”. In graphs, independent variable is positioned in x-axis (horizontal).
Inductive reasoning: Examining specific, individual cases in order to draw a general conclusion. In this regard, inductive reasoning looks for consequences and involves “forward thinking” and projects future outcomes. “What if X situation happens?” is the major question in inductive reasoning. PRA, PHA, FMEA, FMECA, ETA, and HAZOP are inductive risk assessment tools.
Informed consent: A process by which a subject voluntarily confirms his or her willingness to participate in a particular trial, after having been informed of all aspects of the trial that are relevant to the subject’s decision to participate. Informed consent is documented by means of a written, signed and dated informed consent form. (ICH E6/R1)
Prior to a subject’s participation in the trial, the written informed consent form should be signed and personally dated by the subject or by the subject’s legally acceptable representative, and by the person who conducted the informed consent discussion. By signing the consent form, the witness attests that the information in the consent form and any other written information was accurately explained to, and apparently understood by, the subject or the subject’s legally acceptable representative, and that informed consent was freely given by the subject or the subject’s legally acceptable representative.
Both the informed consent discussion and the written informed consent form and any other written information to be provided to subjects should include explanations of the following:
- That the trial involves research.
- The purpose of the trial.
- The trial treatment(s) and the probability for random assignment to each treatment.
- The trial procedures to be followed, including all invasive procedures.
- The subject’s responsibilities.
- Those aspects of the trial that are experimental.
- The reasonably foreseeable risks or inconveniences to the subject and, when applicable, to an embryo, fetus, or nursing infant.
- The reasonably expected benefits. When there is no intended clinical benefit to the subject, the subject should be made aware of this.
- The alternative procedure(s) or course(s) of treatment that may be available to the subject, and their important potential benefits and risks.
- The compensation and/or treatment available to the subject in the event of trial-related injury.
- The anticipated prorated payment, if any, to the subject for participating in the trial.
- The anticipated expenses, if any, to the subject for participating in the trial.
- That the subject’s participation in the trial is voluntary and that the subject may refuse to participate or withdraw from the trial, at any time, without penalty or loss of benefits to which the subject is otherwise entitled.
- That the monitor(s), the auditor(s), the IRB/IEC, and the regulatory authority(ies) will be granted direct access to the subject’s original medical records for verification of clinical trial procedures and/or data, without violating the confidentiality of the subject, to the extent permitted by the applicable laws and regulations and that, by signing a written informed consent form, the subject or the subject’s legally acceptable representative is authorizing such access.
- That records identifying the subject will be kept confidential and, to the extent permitted by the applicable laws and/or regulations, will not be made publicly available. If the results of the trial are published, the subject’s identity will remain confidential.
- That the subject or the subject’s legally acceptable representative will be informed in a timely manner if information becomes available that may be relevant to the subject’s willingness to continue participation in the trial.
- The person(s) to contact for further information regarding the trial and the rights of trial subjects, and whom to contact in the event of trial-related injury.
- The foreseeable circumstances and/or reasons under which the subject’s participation in the trial may be terminated.
- The expected duration of the subject’s participation in the trial.
- The approximate number of subjects involved in the trial.
Inspection: The act by a regulatory authority(ies) of conducting an official review of documents, facilities, records, and any other resources that are deemed by the authority(ies) to be related to the clinical trial and that may be located at the site of the trial, at the sponsor’s and/or contract research organization’s (CRO’s) facilities, or at other establishments deemed appropriate by the regulatory authority(ies). (ICH E6/R1)
Installation qualification (IQ): The process of obtaining and documenting evidence that the premises, equipment and supporting systems have been provided and installed in compliance with their design specifications. (WHO)
Institutional Review Board (IRB): An independent body constituted of medical, scientific, and non-scientific members, whose responsibility is to ensure the protection of the rights, safety and well-being of human subjects involved in a trial by, among other things, reviewing, approving, and providing continuing review of trial protocol and amendments and of the methods and material to be used in obtaining and documenting informed consent of the trial subjects. (ICH E6/R1)
Instrument: A device that interprets a mechanical, digital or analogue signal generated by a sensor, and converts it into engineering units (e.g., °C, percentage relative humidity, mA) through scaling.
Insulated shipper: A single-use insulated passive container, containing coolant, typically used to distribute TTSPPs by road or air transport. (WHO)
Insulated shipping container: Type of packaging used to ship time and temperature sensitive pharmaceutical product (as well as perishables and chemicals). (WHO)
EPS puzzle based insulated shipping container (Mediline Isothermal Solutions)

Intention-to-treat principle: The principle that asserts that the effect of a treatment policy can be best assessed by evaluating on the basis of the intention to treat a subject (i.e., the planned treatment regimen) rather than the actual treatment given. It has the consequence that subjects allocated to a treatment group should be followed up, assessed and analyzed as members of that group irrespective of their compliance to the planned course of treatment. (ICH E9)
Interim clinical trial/study report: A report of intermediate results and their evaluation based on analyzes performed during the course of a trial. (ICH E6/R1)
Intermediate vaccine store: A secondary store or substore that receives vaccine either from a primary vaccine store or another intermediate vaccine store and distributes vaccine to lower levels. (WHO)
Intermediates: Material produced during the manufacturing process, which is not yet in the final product, but whose manufacture is critical for the successful production of the actual vaccine. As part of quality assessment, both quantifiable and qualitative parameters of an intermediate should be defined and specifications established to determine the successful completion of the manufacturing step prior to continuation of the manufacturing process. This includes material that may undergo further molecular modification or be held for an extended period of time prior to further processing. (WHO)
Internal distribution: Transport of a TTSPP within a pharmaceutical manufacturer’s internal supply chain (i.e., all internal transport from the manufacturing plant to the packaging plant and onwards to warehouses and distribution centres). Contrast with external distribution. (WHO)
International Clinical Trials Registry Platform (ICTRP):  A global initiative aiming to make information about all clinical trials involving human beings publicly available. It was established in 2006 in response to demand from countries through the World Health Assembly for “a voluntary platform to link clinical trials registers in order to ensure a single point of access and the unambiguous identification of trials with a view to enhancing access to information by patients, families, patient groups and others". The Secretariat of the ICTRP is housed by the World Health Organization in its headquarters in Geneva. WHO regards trial registration as the publication of an internationally-agreed set of information about the design, conduct and administration of clinical trials. These details are published on a publicly-accessible website managed by a registry conforming to WHO standards. For further details visit http://www.who.int/ictrp/en/
A global initiative aiming to make information about all clinical trials involving human beings publicly available. It was established in 2006 in response to demand from countries through the World Health Assembly for “a voluntary platform to link clinical trials registers in order to ensure a single point of access and the unambiguous identification of trials with a view to enhancing access to information by patients, families, patient groups and others". The Secretariat of the ICTRP is housed by the World Health Organization in its headquarters in Geneva. WHO regards trial registration as the publication of an internationally-agreed set of information about the design, conduct and administration of clinical trials. These details are published on a publicly-accessible website managed by a registry conforming to WHO standards. For further details visit http://www.who.int/ictrp/en/
International Federation of Pharmaceutical Manufacturers and Associations
(IFPMA):  Founded in 1968, the IFPMA is a global, non-profit, nongovernmental organization with members across the globe and a secretariat based in Geneva, Switzerland. The IFPMA represents the research-based pharmaceutical industry, including the biotechnology and vaccine sectors. The IFPMA advocates policies that encourage discovery of and access to life-saving and life-enhancing medicines to improve the health of people everywhere. For details visit http://www.ifpma.org/
Founded in 1968, the IFPMA is a global, non-profit, nongovernmental organization with members across the globe and a secretariat based in Geneva, Switzerland. The IFPMA represents the research-based pharmaceutical industry, including the biotechnology and vaccine sectors. The IFPMA advocates policies that encourage discovery of and access to life-saving and life-enhancing medicines to improve the health of people everywhere. For details visit http://www.ifpma.org/
International Medical Products Anti-Counterfeiting Taskforce (IMPACT): A taskforce administered by WHO in 2006, aiming to build coordinated networks across and between countries in order to halt the production, trading and selling of fake medicines around the globe. IMPACT is a partnership comprised of all the major anti-counterfeiting players, including: international organizations, non-governmental organizations, enforcement agencies, pharmaceutical manufacturers associations and drug and regulatory authorities. (WHO)
International nonproprietary name (INN): An official generic name given to a pharmaceutical drug or active ingredient. INN facilitates the identification of pharmaceutical substances or active pharmaceutical ingredients. Each INN is a unique name that is globally recognized and is public property. A nonproprietary name is also known as a generic name. (WHO)
International Pharmacopoeia (Ph. Int.): The International Pharmacopoeia (Ph. Int.) is published by WHO with the aim to achieve a wide global harmonization of quality specifications for selected pharmaceutical products, excipients and dosage forms. The activities related to Ph.Int. are an essential element in overall quality control and assurance of pharmaceuticals contributing to the safety and efficacy of medicines. The first volume of Ph.Int. was published in 1951. (WHO)
International standards for clinical trial registers: Standards developed as part of the programme of work of the World Health Organization’s International Clinical Trials Registry Platform. The specific, detailed standards further define the requirements of each registry criterion. The standards were considered by the registries participating in the 1st Meeting of the ICTRP Registry Network held in WHO Headquarters, Geneva, Switzerland 11-12 November 2010, and then finalized. See full text at http://www.who.int/iris/bitstream/10665/76705/1/9789241504294_eng.pdf?ua=1
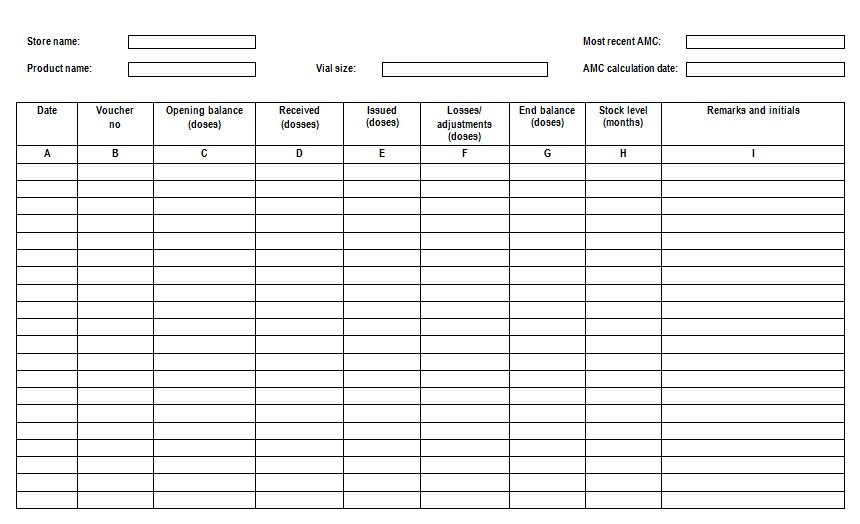
Inventory control card: An individual stock keeping card that keeps information about all batches of a product. One inventory control card should be kept for each product. For example, for a DTP vaccine of three different batches, there should be three batch cards and one inventory control card. In the inventory control card, total quantity in hand of DTP vaccines, as well as the total losses and adjustments regardless of the batch numbers and locations of the products will be seen. Inventory control card is a summary of the batch cards for a product. (WHO)
A sample inventory control card (WHO)
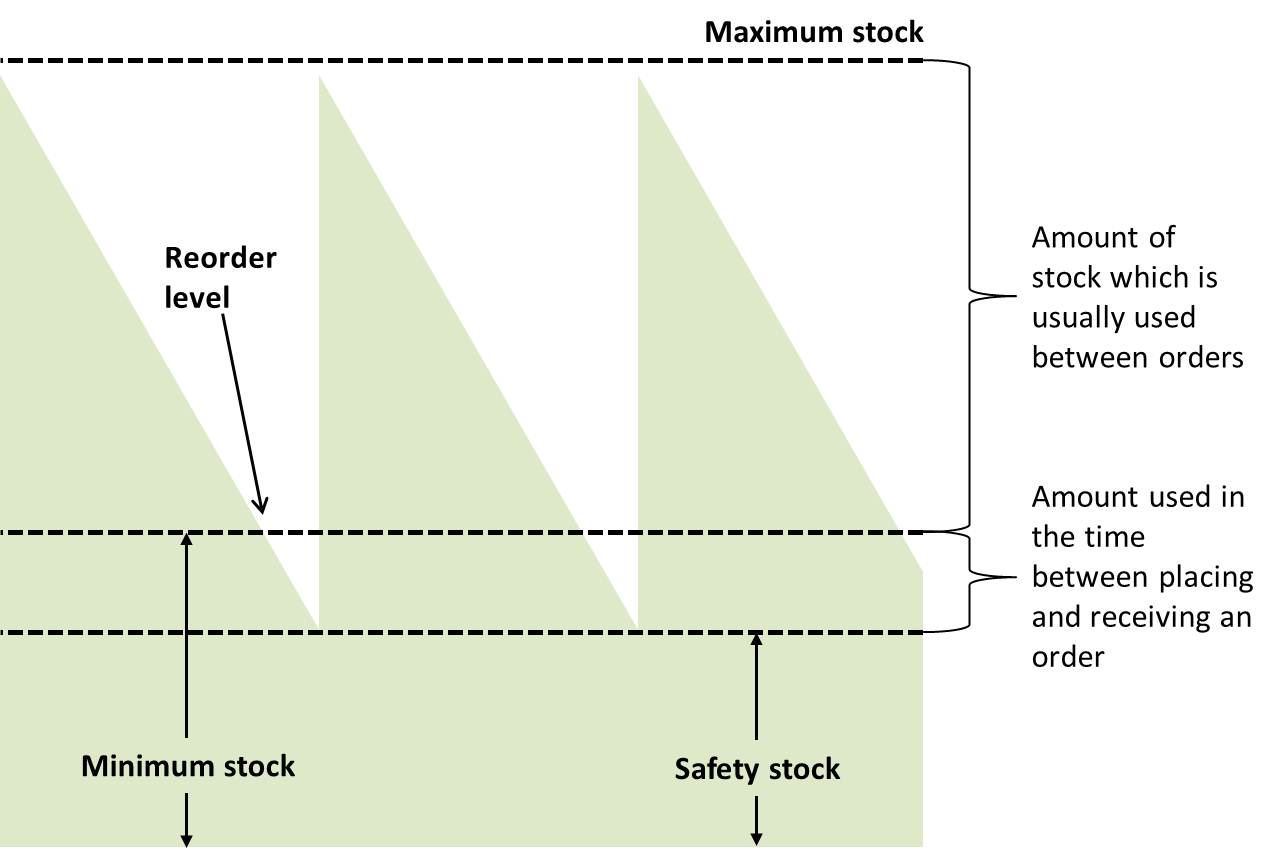
Inventory control system: A process for managing and locating objects or materials. Minimum/maximum (min/max) inventory control system is recommended in pharmaceutical stock management in which, each organizational level of the programme is assigned maximum and minimum levels for its supplies. Using a min/max inventory control system will help managers to prevent both over-stocking (which leads to higher wastage) and shortages or stock outs of pharmaceutical product and other related supplies. (WHO)
Inventory turnover: A measure of the number of times inventory is sold or used in a time period such as a year. The equation for inventory turnover equals the cost of goods sold divided by the average inventory. Inventory turnover is also known as inventory turns, stockturn, stock turns, turns, and stock turnover. (WHO)
Stock movement and relation between minimum, maximum and safety stock levels (WHO)
Investigational product: A pharmaceutical form of an active ingredient or placebo being tested or used as a reference in a clinical trial, including a product with a marketing authorization when used or assembled (formulated or packaged) in a way different from the approved form, or when used for an unapproved indication, or when used to gain further information about an approved use. (ICH E6/R1)
Investigator: A qualified scientist who undertakes scientific and ethical responsibility, either on his/her own behalf or on behalf of an organization/ firm, for the ethical and scientific integrity of a research project at a specific site or group of sites. In some instances a coordinating or principal investigator may be appointed as the responsible leader of a team of sub-investigators. The investigator(s) should be qualified by education, training, and experience to assume responsibility for the proper conduct of the trial, should meet all the qualifications specified by the applicable regulatory requirement(s), and should provide evidence of such qualifications through up-to-date curriculum vitae and/or other relevant documentation requested by the sponsor, the IEC, and/or the regulatory authority(ies). (WHO) See also sub-investigator.
Investigator’s brochure (IB): A compilation of the clinical and nonclinical data on the investigational product(s) that are relevant to the study of the product(s) in human subjects (ICH E6/R1). Its purpose is to provide the investigators and others involved in the trial with the information to facilitate their understanding of the rationale for, and their compliance with, many key features of the protocol, such as the dose, dose frequency/interval, methods of administration, and safety monitoring procedures. The IB also provides insight to support the clinical management of the study subjects during the course of the clinical trial. ICH section 7 of the E6/R1 Guideline for Good Clinical Practice (Current Step 4 version, dated 10 June 1996) delineates the minimum information that should be included in an IB and provides suggestions for its layout. Generally, the sponsor is responsible for ensuring that an up-to-date IB is made available to the investigator(s) and the investigators are responsible for providing the up-to-date IB to the responsible IECs.
Example of title page and table of contents for investigator brochure as suggested by the ICH E6/R1


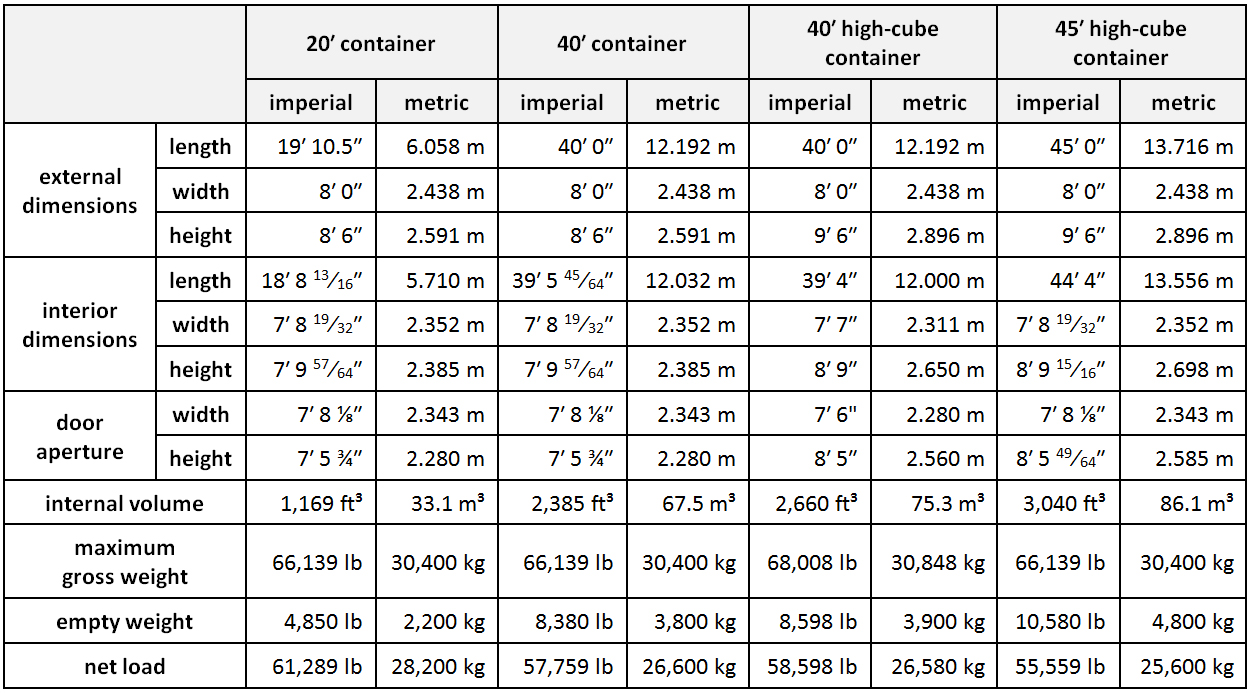
ISO container: Large standardized shipping container, designed and built for intermodal freight transport, meaning these containers can be used across different modes of transport - from ship to rail to truck - without unloading and reloading their cargo. Intermodal containers are primarily used to store and transport materials and products efficiently and securely in the global containerized intermodal freight transport system, but smaller numbers are in regional use as well. These containers are known under a number of names, such as simply container, cargo or freight container, shipping, sea or ocean container, container van or Conex box.
A 20-foot (6.09 m) long shipping container. Each of the eight corners has an essential twist lock fitting for hoisting, stacking, and securing (Eugene Sergeev, Shutterstock)

A few relevant ISO series standards include:
- ISO 6346:1995 Freight containers - Coding, identification and marking
- ISO 668:2013 Series 1 freight containers - Classification, dimensions and ratings
- ISO 1161:1984 Series 1 freight containers - Corner fittings - Specification
- ISO 1496-1:2013 Series 1 freight containers - Specification and testing - Part 1: General cargo containers for general purposes
Weights and dimensions of the most common standardized types of containers are given below. Values vary slightly from manufacturer to manufacturer, but must stay within the tolerances dictated by the standards.
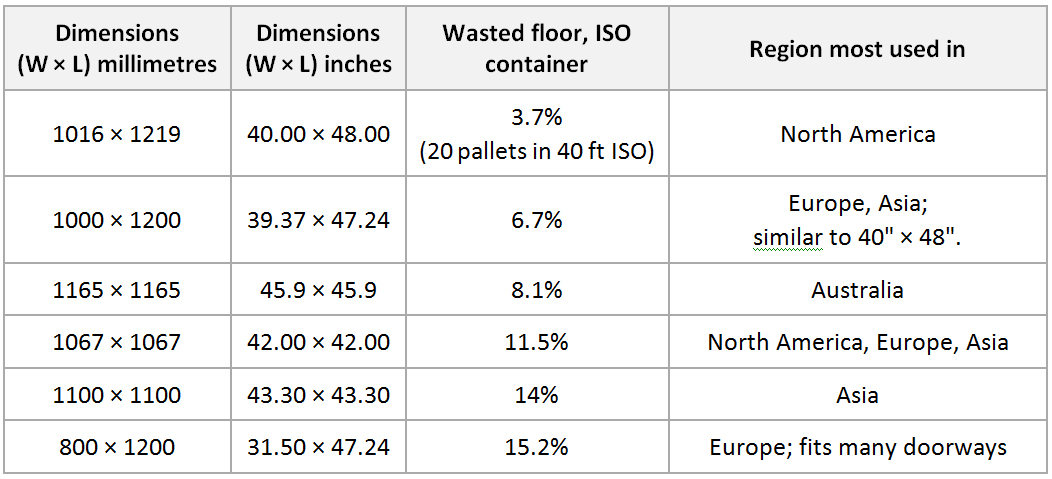
ISO pallets: The International Organization for Standardization (ISO) sanctions six pallet dimensions, detailed in ISO Standard 6780: Flat pallets for intercontinental materials handling—Principal dimensions and tolerances:
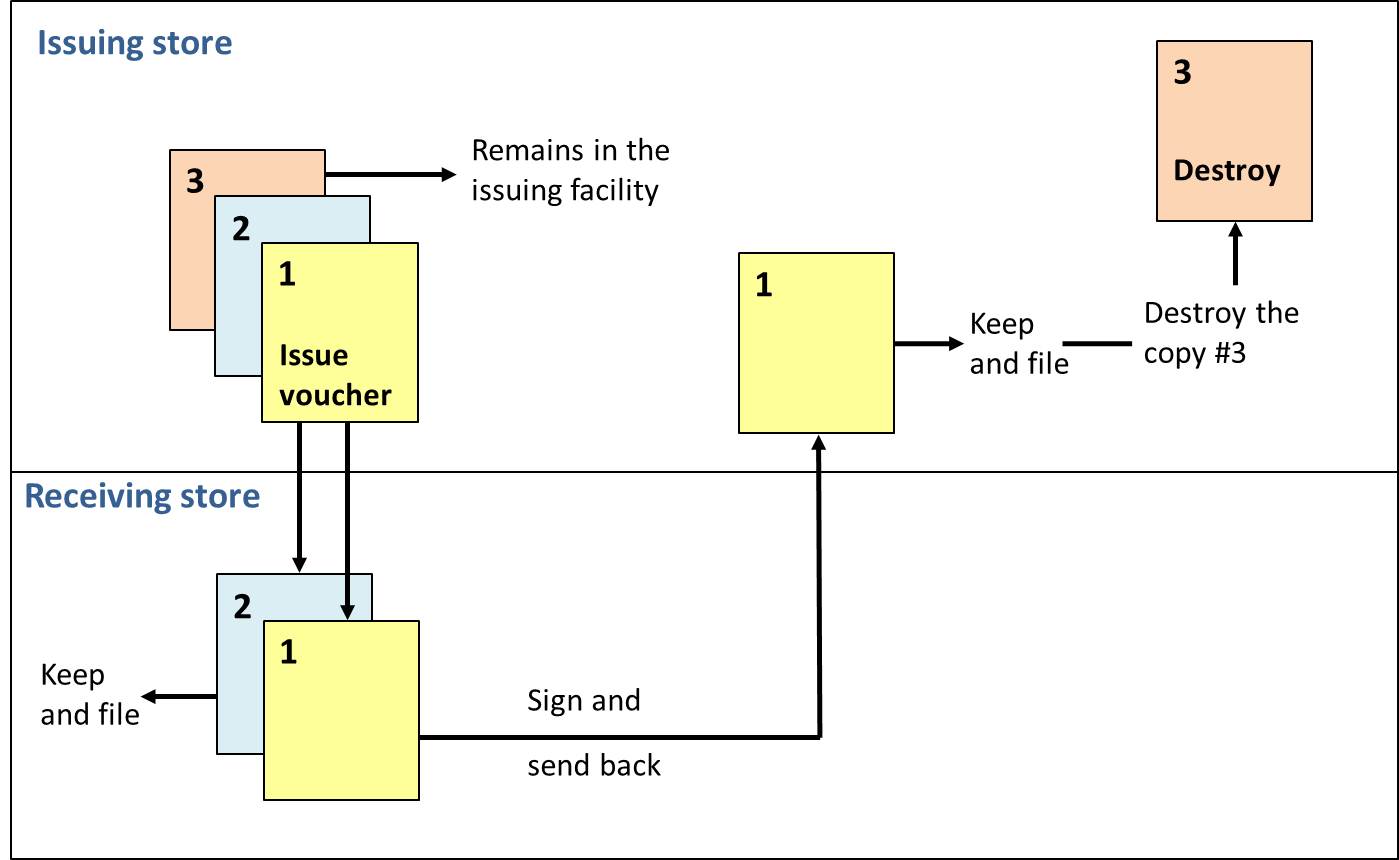
Issue voucher: Transaction record that lists the items and quantities of products issued to a facility.
A sample issue voucher (WHO)
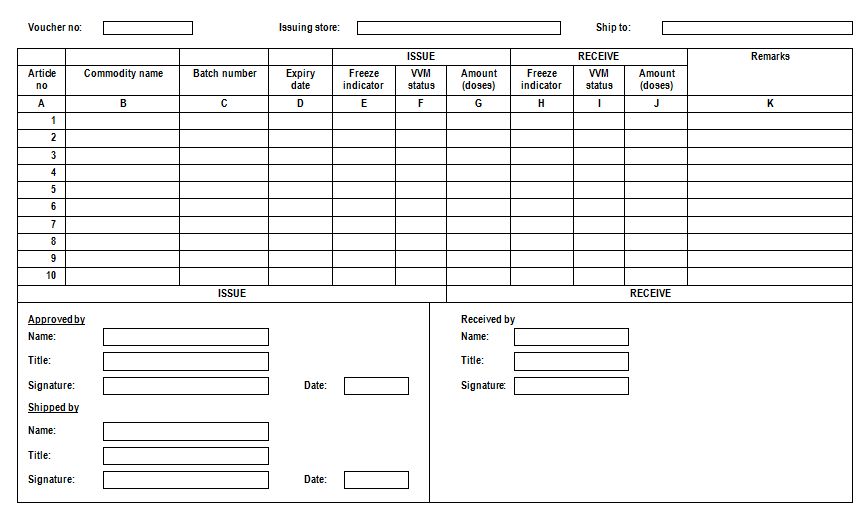
An issue voucher should be completed in three copies. The issuing facility completes the date and quantities issued, signs the record, and sends the top two (1 and 2) copies to the receiving facility along with the supplies. The bottom copy (3) is remained in the issuing facility. The receiving facility verifies the quantity received, signs the form, and sends the top copy (1) back and keeps the middle copy (2) for its records. The top copy (1) arrives at the issuing facility, which issuing facility then disposes of copy (3) and keeps the top copy (1) for its files. At the end, each of the facilities ends up with a completed copy of the issue voucher for filing (see figure).
Flow diagram of issue voucher (Kartoglu)
Issues data: Information on the quantity of goods shipped from one level of a system to another. See also consumption data.


The users of this electronic publication are free to share (to copy, distribute, display and perform the work and make derivative works based on it only for noncommercial purposes); and to remix (to adapt the work) under the following conditions:
Attribution - The work must be attributed in the manner specified by the author or licensor (but not in a way that suggests that they endorse you or your use of work)